










Alberto Kornblihtt lleva más de un cuarto de siglo investigando el funcionamiento del splicing alternativo, lo que él llama “el sastre de nuestras células”. En particular, su grupo de trabajo en el IFIBYNE indaga en la especificidad de un mecanismo clave: cómo la velocidad de la transcripción del ADN a ARN mensajero regula el splicing alternativo. La publicación en Cell de este último paper producido por su equipo vuelve a poner de relieve que no se puede obtener ciencia aplicada sin una perseverante y sostenida investigación en ciencia básica que explique, antes, la íntima maquinaria molecular de nuestro organismo, para entonces sí aspirar a curarlo.
Lo que sigue es un sucinto y más o menos didáctico resumen de los fundamentos, edificados pacientemente a lo largo de años, sobre los que se generó este aporte al tratamiento de una enfermedad tan devastadora como la atrofia muscular espinal que, desde luego, no estaba entre los objetivos iniciales de Kornblihtt y su equipo, pero llega como resultado de una incesante tarea de construcción de conocimiento.
Primero, ¿qué es el splicing alternativo? El principal producto de los genes son las proteínas. ¿Cómo se fabrican? La información genética de todas las células está contenida en la molécula de ADN. Hay porciones del ADN que pueden ser transcriptas para formar moléculas de ARN, y algunos de esos ARN, los llamados “mensajeros”, pueden tener un segundo copiado o traducción; los genes así transcriptos por una enzima dan origen a las proteínas.
Ahora bien, el ADN está hecho de piecitas llamadas exones, interrumpidas por otras intercaladas, los intrones. Cuando la enzima copia el gen, fabrica moléculas de ARN “inmaduro” que contienen información tanto de los exones como de los intrones. Allí, en el núcleo de la célula, se conforma una partícula, el espliceosoma, que se ocupa de eliminar los intrones y unir los exones. Corta y empalma. Se “cose” de este modo un ARN mucho más corto que el gen original. El hallazgo y la descripción de este fenómeno, conocido como splicing, les valió a Phillip Sharp y Richard Roberts un Nobel de Medicina en 1993.
Al poco tiempo se descubrió el splicing “alternativo”, que resultó ser no la excepción sino la regla. Al ser transcripto un mismo gen y ocurrir el splicing, en algunas moléculas se eliminan sólo los intrones y en otras se elimina también algún exón que está en el medio de dos intrones. Se producen así dos variantes de ARN mensajero, con y sin el exón alternativo. Por lo tanto, ese gen puede fabricar dos proteínas similares pero no idénticas. Miles, en realidad. El splicing alternativo explica la vasta diversidad de proteínas que se genera a partir de un número limitado de genes.
Aparece aquí la metáfora del “sastre”. Como suele explicar Alberto Kornblihtt, si el ARN mensajero precursor es una tela, el espliceosoma corta y cose en distintos puntos de esa tela, incluyendo o excluyendo este o aquel exón, y fabrica entonces, al partir del mismo gen, distintos “trajes”, distintas proteínas.
Las mutaciones que alteran las secuencias regulatorias del splicing alternativo son frecuentes en las enfermedades hereditarias y en el cáncer. Y para desentrañar esos fenómenos es clave comprender la maquinaria de la transcripción, que ocurre de modo simultáneo: a medida que la enzima avanza por el ADN fabricando ARN, también actúa el espliceosoma cortando y uniendo. ¿Y cuál es una de las variables fundamentales que regulan las opciones de exclusión/inclusión de exones del splicing alternativo? La velocidad de la transcripción.
En el 80% de los fenómenos, si la velocidad de la polimerasa –la enzima que transcribe el ADN– es baja, permite una mayor inclusión de exones alternativos en el ARN mensajero maduro. En el 20% de los casos, por el contrario, la inclusión del exón alternativo es mayor cuanto más rápido es el avance de la enzima. Hay condiciones fisiológicas y patológicas que generan cambios en la propia enzima, que la ralentan o aceleran. Pero el objeto de estudio de Kornblihtt y su equipo es la cromatina.
La cromatina es la forma en que está empaquetado el ADN, asociado a proteínas llamadas histonas. El grado de compactación de la cromatina puede facilitar o dificultar el pasaje de la enzima que transcribe, controlar su velocidad y, por lo tanto, modificar el splicing alternativo. Si el ADN presenta una cromatina laxa, la polimerasa va más rápido. Si la cromatina está compacta, la polimerasa va más lento y, en general, hay una mayor inclusión del exón alternativo. Por fin, hay dispositivos químicos que permiten regular esa compactación. Si las histonas están acetiladas, la cromatina se abre (y la velocidad de la transcripción aumenta). Si están metiladas, por el contrario, se compacta.
¿Cómo se relaciona este mecanismo con la atrofia muscular espinal? La atrofia muscular espinal (AME) es la primera enfermedad neurodegenerativa que se trata a partir de conocimientos básicos sobre splicing. En los casos más graves, las neuronas motoras que inervan a los músculos no funcionan, los músculos no se contraen, tampoco los intercostales o el diafragma, y sin ventilación mecánica externa, el paciente muere.
El gen mutado en esta enfermedad es el SMN1. Por alguna razón aun desconocida, las neuronas motoras son muy sensibles a la merma en la cantidad de proteína que fabrica este gen. Si el SMN1 está mutado, la proteína no se fabrica. Pero ese gen tiene un backup, el SMN2, que difiere en 11 nucleótidos del SMN1 y es el que permite que, aun con la enfermedad, el niño nazca.
De nuevo, la razón es el splicing alternativo: uno de esos cambios en los nucleótidos del SMN2 permite que actúe un mecanismo inhibidor de la inclusión del exón 7 en el ARN mensajero maduro. Sin el exón 7, el splicing produce un 80% de proteína enferma. Con ese exón incluido, el 20% de la proteína es sana y por eso el embrión llega a término.
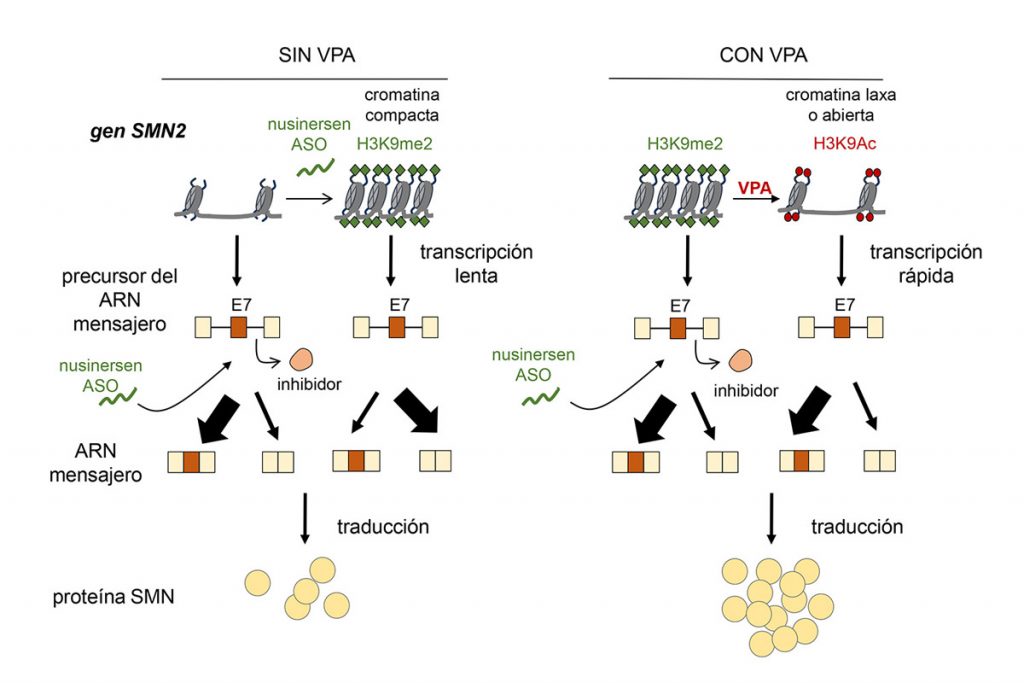
El ácido valproico (VPA) inhibe la desacetilación de las histonas y revierte el efecto de metilación que compacta la cromatina, producido por la acción del oligonucleótido (nusinersen). Esto hace que la transcripción del ARN mensajero se acelere, favoreciendo la inclusión del exón 7 del gen SMN2 en el splicing alternativo, lo que le permite fabricar más proteína sana.
Con esta información, el biólogo molecular uruguayo Adrián Krainer desarrolló en los Estados Unidos una estrategia poco convencional. En lugar de intentar sanar el SMN1, procuró intervenir sobre el splicing alternativo del SMN2: despegar ese inhibidor para que el gen backup fabrique mucha proteína sana. Diseñó entonces un oligonucléotido sintético, un trozo de ADN que desplaza ese inhibidor, aumentando la inclusión del exón alternativo. Eso es Spinraza, una droga aprobada a fines de 2016 para el tratamiento de la atrofia muscular espinal.
Para entonces, un grupo de familiares de niños con AME se acercó a Kornblihtt. ¿Podía hacer su equipo algún aporte a esa investigación? Kornblihtt no quería venderles “espejitos de colores”, les pidió tiempo para saber si había, realmente, una hipótesis de trabajo que pudieran desarrollar. Luciano Marasco se puso la investigación al hombro, y al fin lograron responder una pregunta clave: la inclusión del exón 7 del gen SMN2 ¿está regulada por la velocidad de la transcripción? La respuesta es «sí».
De hecho, corroboraron que la inclusión de ese exón está regulada por aquel modelo del 20% de eventos en los que, si la transcripción es lenta, el exón 7 se incluye poco. Necesitaban acetilar las histonas (más precisamente, inhibir su desacetilización), abrir la cromatina, acelerar el paso de la polimerasa en la transcripción, favorecer la inclusión del exón en el splicing alternativo, que el gen produjera más proteína sana. Funcionó.
Contado así, en pocas líneas, parece sencillo, pero no hay atajos en la investigación en ciencia básica. Hay un largo camino de esfuerzo y dedicación y, eventualmente, resultados como éste, que brindan una esperanza a cientos de familias.



Alvaro Figueroa
9 June 2022 - 14:30
Excelente nota, ojalá se mantenga un flujo permanente de información científica de calidad en este diario